
One question that sometimes comes up regarding DFT computation (typically at zero pressure) is whether one can safely neglect the effect of pressure at ambient conditions. Here’s a simple back-of-the-envelope calculation that shows why it’s OK to neglect pressure under normal circumstances.
The effect of pressure on absolute energy
Under constant temperature and pressure conditions, the relevant thermodynamic potential is the Gibbs Free Energy, defined as:
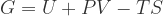
where G is the Gibbs free energy, U is internal energy, P is pressure, V is volume, T is temperature and S is entropy. Since we’re doing a back-of-the-envelope calculation and want to single out pressure effects, let’s conduct our analysis at zero temperature.[1] Also let’s normalize extrinsic quantities per atom; the “per atom” versions will be denoted by lowercase letters:
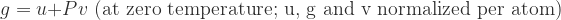
Clearly, the difference between g at finite pressure and zero pressure is the Pv term:
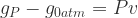
The value of P under ambient conditions is 100 kPa (105 Pa). For v (the volume per atom), let’s plug in values for Si, which has a 40 Angstrom3 unit cell containing 2 atoms,[2] so v ~ (20)*10-30 m3. So:
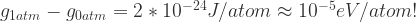
10-5 eV/atom is very small. For comparison, energy differences between different crystal structures are on the order of 10-2 eV/atom. The effect of ambient pressure on absolute energy is about 1000 times smaller than the quantities we care about! Note that the only real assumption in this analysis – other than zero temperature – was v, and this will not vary by too much between different compounds.
The effect of pressure on relative energies between phases
What matters physically is not absolute energy but relative energies between compounds. In particular, at high pressure compounds with smaller v (more dense) will have lower g and thus be preferred.
We can approximate the difference in g between two compounds due to pressure as:
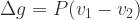
All we need to evaluate this expression numerically are number densities for two different phases. Let’s choose two phases of Si – cubic ground state (v1 ~20 *10-30 m3) and high pressure (v2 ~14*10-30 m3).[2] Then:
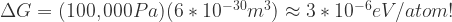
Again, the effect of ambient pressure is several orders of magnitude smaller than what we care about.
Of course, one could always crank up the pressure – a lot. For example, the high-pressure phase of Si is calculated to be about 0.3 eV/atom higher in energy than the ground state (according to DFT-GGA),4 making it quite unstable under ambient conditions. However, according to our calculation above, if we crank up the pressure to about 100,000 times ambient conditions,[3] the effect of pressure would be just enough to overcome the calculated energy difference. Nature agrees – Si is known to transition to the high-pressure form at 112,000 times pressure compared to ambient conditions.[4] So pressure can certainly have an effect in extreme conditions.
The effect of pressure on gases
The assumption that V is about the same order of magnitude for all compounds breaks down for gases. Fortunately, the product PV for a gas can be evaluated using the ideal gas law:
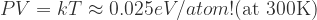
Interestingly, the effect of pressure on the Gibbs free energy depends strongly on the temperature. Note that in contrast to solids, the effect of pressure on gases is large enough that it should be added to the calculations even in normal situations.
References
[1] Note that pressure can have an effect on S; a fundamental thermodynamic relation equates 

[2] I’m using experimental volumes for v at ambient pressure; they’re generally very close to DFT values. Data is from the Materials Project. Ground state (cubic) is mp-149, high pressure (beta-Sn) is mp-92.
[3] Note: to keep the math simple and easy for everyone to follow in their head, I was a bit sloppy with the rounding. If you follow the math more closely you would predict a transition closer to 75,000 times ambient pressure.
[4] Reported in “Phases of Silicon at High Pressure” by Hu and Spain.
